 Faculty of Sciences
Faculty of Sciences (ES)")
")
Fecha: 19 de mayo de 2022.
Hora: 12:30 a 13:30.
Lugar: Aula B01.
Ponente: Prof. Matej Kanduc (Jožef Stefan Institute, Ljubljana, Eslovenia).
Adsorption of amphiphilic molecules to aqueous interfaces occurs in many technological and biological settings, sometimes desired (stabilization of foams), while other times not (contamination). I will discuss how atomistic computer simulations can help us elucidate molecular mechanisms of surfactant adsorption, ranging from short-chain alcohols to double-tail lipids.
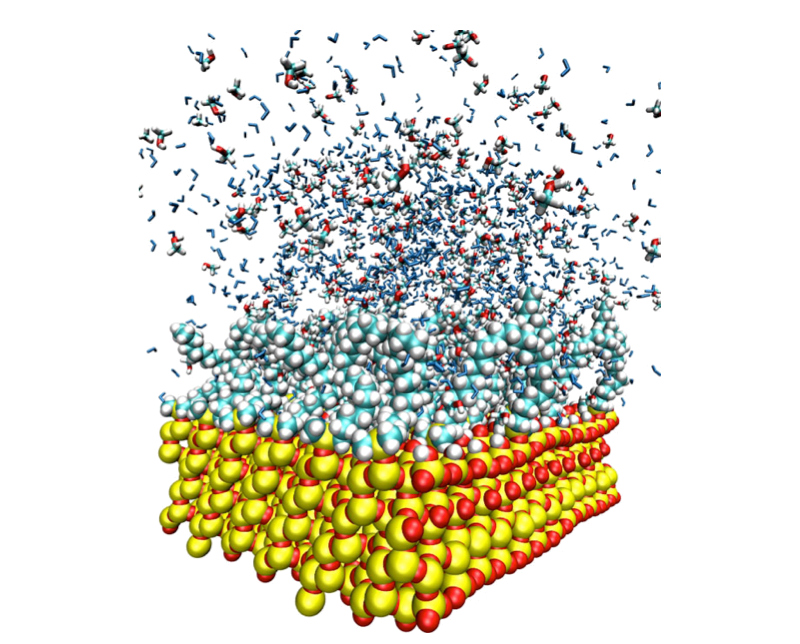
Small surfactants form loose monolayers and exhibit rapid exchange between the interface and bulk, which can be followed in the simulations. Surfactants with longer alkyl chains adsorb as denser monolayers and exchange on experimental timescales that are too slow to be captured in simulations, which challenges molecular modeling. The modeling of this regime requires advanced computational techniques that connect interface and bulk phases via precise determinations of their chemical potentials. Finally, double-chain surfactants (also called "lipids") do not exchange between bulk and interface both on experimental and simulation timescales. They exist as bilayer aggregates already in solution and adsorb to surfaces in the form of monolayers only under certain conditions. As it turns out, a universal determiner for adsorption is the wetting contact angle of the surface. Only surfaces with contact angles larger than around 60-70° are capable of forming lipid monolayers.
Organiza el Grupo de Física de Fluidos y Biocoloides de la UGR.

